
Бесплатный фрагмент - Специфика взаимодействия тонкого и наноуровней микроструктурной организации веществ и их влияние на свойства материалов
Монография

Введение
Машиностроение является одной из наиболее материалоемких отраслей промышленности, которая характеризуется широкой номенклатурой применяемых металлических и неметаллических материалов, а также изготавливаемых из них изделий и конструкций.
Полимеры занимают одно из ведущих мест среди конструкционных материалов в машиностроении. Так, потребление пластмасс в этой отрасли соизмеримо (по объему) с потреблением стали. Целесообразность использования полимеров в машиностроении определяется, прежде всего, возможностью удешевления продукции, экономии металла, в том числе благодаря уменьшению расходов при переработке его в изделия и существенному повышению коэффициента использования. Из таких пластических масс как полиэтилен, фторопласты, полиарилаты, фенопласты, волокниты, стеклопластики изготавливают обширный ассортимент деталей и узлов машин, а также технологическую оснастку различного назначения.
В материаловедении традиционно рассмотрение внутренней организации материала начинают с анализа его «тонкой» структуры. Сегодня различают микро- (включающую в себя тонкую (электронно-ядерную и молекулярную) и наноструктуры), мезо- и макроструктуру материала. При этом очевидно, что электронно-ядерная структура является базовой (исходной) для остальных вышеперечисленных, например, разделяя их на металлы и неметаллы.
В отличие от металлов специфика тонкой структуры полимерных материалов характеризуется не только наличием химических связей атомных остовов, но и межмолекулярного взаимодействия (ММВ) макромолекул между собой. Известно, что ММВ влияет на большинство физических и механических свойств полимеров, в частности, температуру стеклования, размягчения и плавления, растворимость, летучесть, поверхностные свойства, совместимость, вязкость расплавов, кристалличность, прочность, текучесть и т. д. При этом ММВ часто трактуется как остаточное, или вторичное, от химического взаимодействия, но их взаимосвязь не исследована.
Известно, что значимость оценки и прогнозирования конечных эксплуатационных свойств материала с целью обеспечения надежности и долговечности последнего является одним из основных условий его эффективного практического использования в конкретном устройстве или механизме. В частности, в настоящее время существуют методы количественной оценки физико-механических свойств полимерных материалов исходя из их химического строения (например, методы Ван Кревелена, Аскадского, Бицерано). Однако отсутствуют системные исследования зависимости физико-механических свойств материалов от типа связи элементов их тонкой структуры.
Таким образом, актуальность разработки подходов, позволяющих количественно оценивать физико-механические свойства полимерных материалов, при этом являющихся доступными для понимания широким кругом специалистов-материаловедов, нетрудоемкими и недорогими, достаточно очевидна и является сегодня важнейшей проблемой теоретического и практического материаловедения.
В рамках предлагаемой работы развивается подход по оценке физико-механических свойств полимерных материалов на основе элементов их тонкой структуры, который должен позволить максимально точно оценивать общий характер изменения их наиболее практически важных физико-механических свойств и дать возможность прогнозировать значения величин данных свойств в новых полимерных материалах.
Исходя из вышесказанного, в настоящей монографии поставлена следующая цель: выявление особенностей взаимодействия элементов тонкой структуры (атомных остовов и фрагментов макромолекул) широко применяемых в машиностроении полимерных материалов и их влияния на физико-механические свойства исследуемых материалов, включая:
Совершенствование методики расчета компонент химических связей в низкомолекулярных, а также высокомолекулярных соединениях, образующих полимерные материалы.
Подтверждение вторичности ММВ от химического взаимодействия и выявление характера влияния компонент химических связей на компоненты ван-дер-ваальсового (ВДВ) ММВ в веществах, образованных низкомолекулярными соединениями, а также высокомолекулярных соединениях и образуемых ими полимерных материалах.
Нахождение зависимости физико-механических свойств от компонент химической связи элементов тонкой структуры материала.
Апробация разработанных подходов, методик и полученных результатов исследования особенностей гомо- и гетероядерного взаимодействия элементов электронно-ядерной структуры широко применяемых в машиностроении материалов на основе низко- и высокомолекулярных соединений с учетом ее влияния на физико-механические свойства соответствующих материалов, имеющих большое значение в промышленности целом, а также в энергетике и машиностроении в частности.
Поставленные в монографии задачи по углубленному исследованию влияния тонкой структуры материала на его свойства отвечают современным тенденциям в развитии материаловедения [1].
Научная новизна исследования состоит в том, что в нем впервые установлено влияние компонент химической связи на величину ВДВ межмолекулярного взаимодействия (определяющих в совокупности специфику тонкого уровня структурной организации полимеров) и далее на ряд физических и механических свойств полимерных материалов, применяемых в энергетике и машиностроении.
Практическая ценность монографии. Опираясь на единую модель химической связи [2,3] элементов электронно-ядерной структуры материала, разработана методика расчета компонент химических связей в низко- и высокомолекулярных соединениях, образующих полимерные материалы. Это позволило связать компоненты химических связей с компонентами ММВ и установить их влияние на некоторые физико-механические свойства полимерных материалов. Таким образом, на основе найденных зависимостей свойств от компонент химической связи полимерных материалов с одинаковой конформацией макромолекулярной цепи была показана возможность оценки и прогнозирования их физико-механических свойств.
Показан характер влияния степеней ковалентности (Ск), металличности (См) и ионности (Си) химических связей на их жесткость и свойства биядерных соединений.
Совокупность полученных данных позволяет говорить о перспективности разрабатываемых подходов и методик для расчета компонент химических связей в низкомолекулярных жидкостях, а также высокомолекулярных соединениях, образующих полимерные материалы, и установления их влияния на энергию ММВ и физико-механические свойства в соответствующих веществах и материалах.
Апробация полученных результатов исследования
Разработанные методики переданы учреждениям и предприятиям, заинтересованным в их практическом применении (ФГУП ЦНИИГеолнеруд и др.), и внедрены в учебный процесс КГЭУ при проведении лекционных и практических занятий по курсу «Современное материаловедение», включая методические указания и контрольные задания для студентов-заочников [4].
Монография состоит из введения, пяти глав, выводов, списка использованной литературы и приложений, в котором приводятся материалы не вошедшие в основные разделы книги.
Во введении обосновывается актуальность темы исследования.
В первой главе проанализированы различные подходы к оценке физико-механических свойств полимерных материалов, показаны их достоинства и недостатки. Рассмотрено состояние вопроса оценки смешанных типов гомо- и гетероядерного химического взаимодействия элементов электронно-ядерной структуры материала (промежуточных между двумя и тремя предельными типами химического взаимодействия соответственно) и способы расчета каждой из трех компонент связи. Рассмотрена роль ММВ в полимерных материалах.
Во второй главе разработана методика, позволяющая рассчитывать распределение электронной плотности (ЭП) в молекулах низкомолекулярных жидкостей, а также звеньях высокомолекулярных соединений, образующих полимерные материалы, через компоненты образующих их химических связей. Описаны квантово-химические подходы для расчета ЭП и энергии ММВ.
В третьей главе исследовано влияние особенностей взаимодействия элементов тонкой структуры в некоторых низко- и высокомолекулярных соединениях, и органических полимерных материалах. Показано влияние компонент гомоядерной химической связи на ее жесткость и энергию ММВ и свойства ряда модельных низкомолекулярных соединений. А также влияние металлической и ионной составляющих гетероядерной химической связи на ее жесткость, энергию ММВ и свойства модельных низкомолекулярных соединений, низкомолекулярных жидкостей и полимерных материалов.
В четвертой главе показано влияние металлической и ионной составляющих гетероядерной химической связи на физико-механические свойства полимерных материалов: полиэтилен высокого давления (ПЭВД), полипропилен (ПП), поливинилиденфторид (ПВДФ), полиизопрен (ПИ), полиэтилентерефталат (ПЭТФ), поликетон (ПК), поливинилфторид (ПВФ), поливинилхлорид (ПВХ), полиакрилонитрил (ПАН), поливиниловый спирт (ПВС) и сополимеры этилена и 1-гексена.
В пятой главе всесторонне исследуется теория в кокорой электронно-ядерный и наноструктурный уровни организации металлических материалов рассматривается, как основа прогнозирования их свойств, усовершенствования технологий придания им новых заданных свойств.
Глава 1. Особенности структурной организации материалов, образованных молекулярными соединениями
1.1. Основные уровни структурной организации материалов
1.1.1. Полимерные материалы
Согласно единой классификации уровней структурной организации материалов, предложенной в работе [5] и уточненной позднее этими же авторами (табл. 1), в полимерах можно выделить следующие структурные уровни [5,6].
I. Микроструктура:
1 подуровень: электронно-ядерный. Данный подуровень является общим для всех материалов и образован элементами, размер которых лежит в диапазоне от ~1 до 5 Å (0,0001—0,0005 мкм): атомными остовами, химическими связями (обобществленными электронами) и точечными дефектами.
Электронно-ядерная структура полимеров описывает расположение атомных остовов и обобществленных электронов в химическом соединении в виде индивидуальной олиго- или макромолекулы. При этом специфика электронно-ядерной структуры в полимерных материалах (в отличие от металлов) заключается в том, в обобществленные электроны характеризуются большей локализацией между ядрами, обеспечивающей образование дискретной частицы — олиго- или макромолекулы.
Атомный остов — ядро с частью электронной оболочки атома, не принимающей участия в образовании химической связи (то есть необобществленные электроны в совокупности с ядром).
Обобществленные электроны — электроны, которые осуществляют химическую связь, возникающую вследствие перекрывания электронных оболочек, и являются общими для связываемых химических остовов.
Точечные дефекты (нульмерные) — дефекты кристаллической решетки, сравнимые с размерами атомных остовов.
2 подуровень: молекулярный. Этот подуровень образован фрагментами макромолекул (атомными группировками), между которыми действуют более слабые (по сравнению с химическими) внутри- и межмолекулярные ван-дер-ваальсовые и водородные связи. Размер элементов, образующих молекулярный подуровень микроструктуры, лежит в диапазоне от ~5 до ~10 Å (0,0001—0,0005 мкм).
Макромолекула — индивидуальное высокомолекулярное химическое соединение, цепеобразующие атомы которого связаны направленными химическими связями, характеризующееся многократным повторением одного или более типов атомов или групп атомов (составных звеньев) в цепи, в количестве, достаточном для проявления образуемым макромолекулами полимером комплекса специфических свойств, который остается практически неизменным при добавлении или удалении одного или нескольких составных звеньев [7].
Олигомолекула — отличается от макромолекулы меньшей степенью полимеризации (обычно не превышающей 100). Комплекс специфических свойств в олигомерах изменяется при добавлении или удалении одного или нескольких составных звеньев его олигомолекулы [7].
Согласно ИЮПАК — Международному союзу теоретической и прикладной химии (IUPAC — International Union for Pure and Applied Chemistry), — полимер определяется как «вещество, состоящее из молекул, характеризуемых многократным повторением одного или более вида атомов или групп атомов (составных звеньев), связанных друг с другом в количествах, достаточных для того, чтобы обеспечить набор свойств, которые не претерпевают значительного изменения при добавлении или удалении одного или нескольких составных звеньев» [8]. То есть, полимер представляет собой совокупность индивидуальных макро- и/или олигомолекул, связанных в полимерную систему посредством ван-дер-ваальсовых или водородных связей.
Полимеры, как правило, относят к веществам, образующим молекулярные кристаллы. Однако, в случае, когда упаковка макромолекул имеет складчатую или фибриллярную конформации, правильнее было бы говорить о ковалентно-молекулярных кристаллах (точнее — ковалентно- (меж- или внутри-) молекулярных), так как вдоль одного из периодов решетки действуют прочные химические, преимущественно ковалентные, связи (образующие электронно-ядерный подуровень микроструктуры), тогда как вдоль двух других периодов решетки действуют более слабые силы межмолекулярного взаимодействия (соответствующие молекулярному подуровню микроструктуры). Примером может служить полиэтилен, макромолекулы которого находятся в складчатой конформации, образуя ламель, и параметры элементарной решетки которого имеют следующие значения: a = 7.40; b = 4.93; c = 2.534 Å [9]. При этом вдоль периода с действуют химические, преимущественно ковалентные, связи, а вдоль периодов a и b — силы Ван-дер-Ваальса.
3 подуровень: наноструктура. К элементам, образующим наноструктуру в полимерах, можно отнести олиго- и макромолекулы, наночастицы, кристаллиты и ламели. Их размер лежит в широком диапазоне ~1 — 1000 нм.
Существует большое разнообразие наночастиц и способов их классификации. Например, по размерности их можно классифицировать на одномерные, характеризующиеся толщиной (пленки, покрытия и т.д.), двухмерные, характеризующиеся двумя размерами (трубки, волокна и т.д.) и трехмерные (трехмерные частицы, полые сферы и т.д.). Наиболее интенсивные исследования направлены на создание углеродных нанотрубок, которые уникальны своей жесткостью, прочностью и электронными свойствами, а также фуллеренов и дендримеров.
Вопрос о классификации кристаллических образований типа кристаллитов и ламелей до конца не решен [10]. Кристаллитами принято считать области трехмерной упорядоченности цепных макромолекул [10]. Также принято считать, что они являются минимальными дискретными элементами любой устойчивой надмолекулярной организации в твердых полимерах [9].
Ламели считаются кристаллическими образованиями более крупного масштаба, характеризующиеся пластинчатой формой [10]. При этом ламель, с одной стороны, можно считать образованной из кристаллитов. Однако вместе с тем ламель также можно рассматривать в качестве первичной надмолекулярной структуры, состоящей из «листов» и «лепестков», представляющих собой грани роста в направлении кристаллографических осей a и b и в свою очередь составленных из сложенных макромолекулярных цепей и упакованных параллельно. При этом одни и те же морфологические формы в одних случаях могут быть относительно независимыми структурными элементами, а в других — нет [9].
Для данного уровня структуры характерны линейные дефекты типа дислокаций в ламелях кристаллических полимеров или дисклинаций в аморфных полимерах.
II. Мезоструктура. Данный уровень структуры полимерных материалов составляют образованные ламелями небольшие аксиалиты, эдриты и сферолиты (размерами до нескольких десятков мкм). Дефектами, характерными для этого структурного уровня, можно, по-видимому, считать поверхностные дефекты типа дислокационных ансамблей.
III. Макроструктура полимеров образована более крупными надмолекулярными образованиями в виде крупных аксиалитов, эдритов и сферолитов размерами от нескольких десятков мкм и выше. Для этого уровня структурной организации полимерных материалов характерны объемные дефекты типа трещин, пор и т. д.
Для металлов можно выделить сходные уровни структурной организации, которые приведены в табл. 1. Из данных табл. 1 видно, что, так как металлы являются немолекулярными веществами, у них отсутствует молекулярный уровень структуры (в отличие от полимеров, образованных высокомолекулярными соединениями).
1.2. Химическое строение вещества
В 1861 году выдающийся русский химик Александр Михайлович Бутлеров на съезде немецких естествоиспытателей и врачей в Германии (г. Шпейер) выступил с докладом «О химическом строении веществ» [11]. В нем А. М. Бутлеров впервые изложил основы своей теории химического строения веществ (структурной теории), основывающейся прежде всего на обобщении значительного фактического материала по структуре органических химических веществ [11,12]. Таким образом, Бутлеров впервые ввел термин «химическое строение», продуманно дал ему определение и сформулировал основные положения своей теории.
Структурная теория химического строения раскрывает свою фундаментальность в последующих обобщениях, сделанных А. М. Бутлеровым [13]:
— молекула реальна и познаваема, строение ее можно и должно выразить единой рациональной (структурной) формулой;
— молекула — не механическая сумма атомов, а новое качественное образование, результат химического взаимодействия, при котором атомы, влияя друг на друга, изменяют свою структуру. В современном понимании атомы в процессе химического связывания превращаются в ядерные центры (или атомные остовы) химического соединения, связанные обобществленными электронами, т.е. химической связью [2,14,15];
— молекула не есть нечто застывшее, она динамична, то есть способна к превращению.
Значимость теории Бутлерова для естествознания трудно переоценить. Ведь именно она впервые сформулировала понятие «химическое строение», понимая под ним качественно новый уровень организации (строения) вещества. И далее формулируется взаимосвязь свойств веществ с их химическим строением: «Химическая натура сложной частицы определяется натурой элементарных составных частиц, количеством их и химическим строением» [16,17].
Сейчас, после введения А. М. Бутлеровым понятия «химическое строение», оно стало более глубоким и точным. Сегодня химическое строение — это не только порядок валентной связи атомов, их взаимное расположение и влияние в химическом веществе, но характер распределения электронной плотности (локализации обобществленных электронов) в межъядерном пространстве между химически связанными атомами, степени обобществления электронов, ковалентности, ионности и металличности связи, её направленность, длина, прочность и так далее [2].
В настоящее время существует ряд методов количественной оценки физико-механических свойств полимеров, на основе их химического строения.
1.3. Подходы к прогнозированию физико-механических свойств полимерных материалов на основе их химического строения
В настоящее время проблема количественной оценки физико-механических свойств полимеров является одной из актуальных. Существует ряд методов количественной оценки физико-механических свойств полимеров, в основе которых лежит химическое строение.
Подход, предложенный Ван Кревеленом и его сотрудниками [18], основан на идее так называемых «групповых вкладов», согласно которой записываются простейшие эмпирические выражения аддитивного типа, причем данная группа, находясь в разных полимерных звеньях, вносит один и тот же вклад в рассчитываемую характеристику (например, в температуру стеклования, плавления и т. д.). Ван Кревелен предложил использовать следующее уравнение:
где F — оцениваемое свойство, Fi — определяет идентичность и вклад функциональной группы i, а ni — соответствует ее содержанию в молекуле.

Это — чисто эмпирический и интуитивный подход, который основан на аддитивных схемах. Как отмечает его автор, этот подход позволяет с хорошей точностью рассчитывать физические свойства многих линейных полимеров [158].
В дальнейшем этод подход получил свое развитие в работах других ученых. Р. О. Корреа, А. С. Телес, Ж. Е. Урике используют метод «групповых вкладов» второго порядка для ими же разработанного для оценки температуры стеклования полимеров [19,20]. Ученые присваивали групповые вклады 923 полимерам из 1018 и установили корреляцию между «групповыми вкладами» и свойствам полимеров, что позволило им рассчитать свойства у оставшихся 95 полимеров.
Для вычисления используют уравнение:

где F — оцениваемое свойство, Nc — общее число функциональных групп в строении полимера, Sc — число пар этих групп, Fi — вклад групп i в свойство F, Gj — вклад пары j в свойство F, ni и mj — количество групп и пар групп.
По мнению ученых, разработавших данный метод, он является простым, численно надежным, и в отличие от метода Ван Кревелена, использует меньшее количество функциональных групп и имеет простую процедуру вычисления, а также отсутствуют коррекционные члены.
Также ведутся работы по совершенствованию методов «групповых вкладов» первого [21,22] и второго порядка [23].
Существенным недостатком методов групповых вкладов является то, что для многих полимеров, особенно новых, параметры групповых вкладов отсутствуют, таким образом, их свойства нельзя рассчитать. Свойства сополимеров также не могут быть оценены.
Другой подход, развиваемый Дж. Бицерано [24], появился совсем недавно, оно основан на так называемых индексах связанности, что на практике свелось к поиску различных корреляций физических свойств со множеством правил, как находить коэффициенты корреляционных зависимостей. Также в уравнение корреляции дополнительно должны быть введены простые корреляционные индексы для специфических групп и структур для улучшения точности предсказаний. Свойства коррелируются в соответствии с уравнением: Свойства = (Σах) + (структурные параметры) + (атомные и групповые коррекционные члены), где а — коэффициенты, с — индексы связанности.
Большинство свойств коррелируются, используя всего четыре индекса связанности: оХ, оХv — нулевого порядка (атомные) и первого порядка (связи) 1Х и 1Хv [24].
Два индекса нулевого порядка атомных связей определяются следующим уравнением:

где Z — атомный номер, Zv — число валентных электронов в атоме, Nн — число атомов водорода связанных с i-м и j-м атомом, которые стоят рядом.
Данный метод предсказывает свойства путем сложения аддитивных вкладов атомов и связей вместо групп.
По мнению автора данного метода, он может предсказывать свойства многих полимеров, которые не позволял метод Ван Кревелена, а также позволяет оценивать свойства сополимеров.
Другое направление в физике и химии полимеров, связанное с количественным анализом влияния химического строения на физико-химические свойства полимеров и предсказанием этих свойств, развиваемый А. А. Аскадским [25—31], является полуэмпирическим. Согласно этому подходу, уравнение для расчета физических свойств получены на основании представлений физики твердого тела, а калибровка метода осуществляется с помощью физических характеристик полимерных стандартов, свойства которых хорошо изучены. В результате параметры уравнений имеют определенный физический смысл. Общим для всех уравнений является суммирование ряда атомных констант, характеризующих вклады в энергию ММВ, энергию химических связей, ВДВ объем и т. д. Таким образом, подход основан на представлении повторяющегося звена полимера в виде набора ангармоничных осцилляторов, которые описывают термическое движение атомов в поле внутри- и межмолекулярных сил, включая слабые дисперсионные, диполь-дипольные взаимодействия, водородные и химические связи [32—34].
Строго говоря, данный подход не может быть назван аддитивным в обычном понимании этого слова, поскольку рассчитываемые свойства не являются аддитивными по отношению к атомам и группам, из которых построено повторяющееся звено полимера.
Аддитивность применяется здесь только к таким характеристикам, которые действительно являются аддитивными (ВДВ объём, молекулярная масса, энергия ММВ и другие).
Как отмечает автор, этот подход позволяет с достаточной точностью оценить многие физические характеристики порядка 60 полимеров. Но при этом отмечает, что точность расчета необходимо повысить.
Однако число свойств, которые могут быть оценены этим методом, меньше, чем методом Ван Кревелена и Бицерано.
По мнению Кынина с сотр. [35] результаты расчетов по методу Ван-Кревелена и методу инкрементов, предложенному Аскадским достаточно близки.
Однако в представленных методах недостаточно полно учтено влияние такой важной характеристики полимеров, как энергия ММВ макромолекул полимера (энергия когезии). Влиянием этого фактора пренебрегают при расчетах плотностей и температурных характеристик полимеров, а при расчете параметра растворимости его влияние учитывается введением энергетических поправок на влияние специфических взаимодействий (например, водородных связей), причем введение таких поправок не имеет строгой регламентации и проводится произвольно.
Кынин с сотр. полагают [35], что достаточно удобной энергетической характеристикой полимеров является параметр растворимости dP. Именно он выступает как связующее звено между химическими свойствами и структурой. Очевидно, что энергия ММВ будет зависеть от природы и количества функциональных групп в элементарном звене полимера, что можно легко показать на примере зависимости параметров растворимости алифатических полиамидов от концентрации функциональных групп.
Также авторам очевидно, что удельная энергия межмолекулярного взаимодействия, а также и связанные с ней плотность энергии когезии и параметр растворимости полимера оказывают существенное влияние на свойства полимеров. Именно от структуры в значительной степени зависят термические, механические и физико-химические свойства материалов [35].
Таким образом, Кыниным с сотр. [35] предлагается физически обоснованный метод оценки изменения физико-химических и физико-механических свойств волокнообразующих полимеров при переменных условиях окружающей среды, изменяющихся в широких пределах, который основан на анализе изменения энергии межмолекулярного взаимодействия в полимере. Использование в качестве характеристики процесса такого универсального показателя, как параметр растворимости, связанный с энергией когезии, позволяет использовать найденные закономерности для новых полимерных материалов и взаимодействия полимеров с любыми низкомолекулярными веществами.
Но, как известно химическое строение вещества определяется, прежде всего, характером (типом) химического связывания атомов и их преобразованием в различные элементы структуры (химические элементы) вещества. Поэтому основу современной теории химического строения составляет учение о химической связи, как основного условия возникновения и существования химического соединения, характеризуемого определенной (химической) структурной организацией [14].
1.4. Тонкая структура материала и ее характеристики
Как следует из табл. 1, тонкий уровень структуры полимерных материалов включает два подуровня: электронно-ядерный и молекулярный. Соответственно под электронно-ядерным подуровнем понимается химическая структура (характер расположения атомных остовов и обобществленных электронов между ними), а под молекулярным — физическая структура (определяемая межмолекулярными взаимодействиями между молекулами низкомолекулярных веществ или фрагментами макромолекул высокомолекулярных соединений).
1.4.1. Характеристика смешанных (промежуточных) типов взаимодействия элементов электронно-ядерной структуры материала
Химическая связь — это совокупность сил, удерживающих нуклиды (ядра) или атомные остовы в химическом соединении. Характеристики химической связи в соединении атомов различных веществ и материалов на их основе определяет его химическую структуру и физико-химические свойства [36]. Различают три предельных вида химической связи: ковалентную, металлическую и ионную. Металлическая связь характерна для металлов, то есть для атомов элементов, характерными свойствами которых являются хорошая теплопроводность, электропроводность, металлический блеск. Для неметаллов характерна ковалентная связь. При взаимодействии неметалла и металла возникает гетероядерная ионная связь.
В зависимости от преобладания в веществе того или иного типа связи существуют три типа химических структур: преимущественно металлическая, преимущественно ионная и преимущественно ковалентная. Элементами металлической структуры являются катионы в узлах кристаллической решётки и обобществлённые электроны между ними. В металле максимальная стабильность структуры связана с максимальным координационным числом. Характерные структуры металлов представляют два типа плотной упаковки элементов с координационным числом, равным 12 и центрированной кубической структуры с координационным числом, равным 14 [37,38].
Элементами ионной структуры служат катионы и анионы в узлах кристаллической решётки, обобществленные электроны которой максимально смещены в сторону электроотрицательного элемента. Ионная связь обеспечивается кулоновским притяжением избыточных электрических зарядов противоположно заряженных ионов. Атомы металлов легко теряют свои внешние электроны, которые стремятся присоединить атомы неметаллов. Таким образом, могут возникнуть стабильные катионы и анионы, которые могут в основном сохранить свои электронные структуры при приближении друг к другу и образовании стабильной молекулы или кристалла [37]. В структуре ковалентных соединений элементами являются пара обобществленных электронов и ядра (в случае водорода — протоны) или атомные остовы, состоящие из ядра и внутренних электронов (в случае остальных атомов) [37].
В работе [39] рассмотрена связь между структурой и свойствами соединений на основе полимеров. Способность элементов образовывать полимеры зависит от таких факторов, как электроотрицательность (ЭО), гибридизация, электронная конфигурация, степень ковалентности (СК) связи между элементами, стерические факторы, координационным числом атомов элементов. Полимерным считается вещество, в котором наличествуют цепи с преимущественно ковалентными связями. Связь между цепями может быть ван-дер-ваальсовой, водородной, ионной, металлической или ковалентной [39].
Гетероцепные полимеры в своей структуре удерживаются в основном двумя типами химических связий преимущественно ковалентной внутри цепей и преимущественно ионной между цепями и лигандами. Исходя из этого все элементы Периодической системы (ПС) подразделяются на каркасообразующие (B, O, Se, S), мостиковые (O) и модифицирующие (H, Li, Na, K, Rb, Cs) элементы. Это зависит от их расположения в ПС и их электронной конфигурации и специфики химической связи [64].
Однако такое разделение на основные предельные типы связи весьма условно. При рассмотрении реальных соединений говорят о смешанном типе химического взаимодействия [40,41]. Однако правильнее считать, что химические связи в реальных молекулах и кристаллах имеет не идеальный — ионный, ковалентный или металлический характер, а какой-то промежуточный. Другими словами, если в ковалентных связях электронная плотность (ЭП) распределена совершенно симметрично между партнёрами и её центр тяжести находится в середине межъядерного расстояния, то в теоретически чисто ионных соединениях он совпадает с центром одного из атомов [42].
Таким образом, из вышеизложенного видно, что тип химической и межмолекулярной связи определяет тонкую структуру вещества, а от нее в свою очередь зависят физические, химические и механические свойства веществ и материалов. Другими словами, тонкий (электронно-ядерный и молекулярный) уровень строения вещества является базовым для всех типов веществ и материалов на их основе и влияет на следующие за ним уровни структурной организации материала.
1.5. Электроотрицательность элементов тонкой электронно-ядерной структуры материала
1.5.1. Шкалы электроотрицательностей
Само понятие электроотрицательность впервые было применено Берцелиусом в 1811 г. для классификации элементов, в 1858 г. ЭО и валентности атомов использовал Канницаро для характеристики элементов, в 1903 г. Штарк предложил определять ЭО атома по энергиям его ионизации и сродства к электрону, наконец, Льюис (1916) в теории ковалентной связи рассматривал полярность как смещение центра тяжести валентного электронного облака в сторону одного из атомов — именно он в данной связи является электроотрицательным. Отсюда следует, что этот параметр отражает способность атома притягивать электроны от связанных с ним атомов (Инголд, 1929 год). В этом ряду блестящих имён Полингу принадлежит честь создания первой количественной шкалы ЭО, основанной на термохимических данных [43].
Таким образом, электроотрицательность отражает способность атомов притягивать к себе электроны. Чем выше электроотрицательность, тем сильнее эта способность [43]. На протяжении более 50 лет концепция электроотрицательности модифицировалась, расширялась. К 1988 году уже стало возможно связать электроотрицательность составных элементов со свойствами сотен соединений, а также вычислить значение энергий полярных ковалентных связей.
Электроотрицательность является мерой эффективного заряда ядра. Сандерсон [44] отмечает, что непосредственной мерой ЭО является не средняя ЭП, а отношение ЭП атома к средней ЭП атома «благородного» газа. Также автор приходит к выводу о чередовании значений ЭО вниз по группе.
Сандерсон критикует систему ЭО Полинга, мотивируя это тем, что полярную связь невозможно представить простым суммированием «100%-ной ковалентности + Х%-ной ионности» и говорит о том, что сумма ионности и ковалетности должна в сумме составлять 100%. Он делает вывод, что «концепция ЭО, как свойства атома доказала свою целесообразность чрезвычайно широким распространением и точным количественным применением» [44], а также о том, что электроны, не участвующие в образовании связи, «уменьшают электроотрицательность исходных элементов, усиливают полярность связи с более ЭО элементами, за счет чего прочность связи возрастает по сравнению с нормальной валентностью» [44].
Концепция ЭО решает задачу количественной характеристики химической связи. Она много критиковалась в свете существующих неэмпирических методов распределения ЭП в молекулах. Однако эта теория оказалась жизнеспособной. В пользу этого свидетельствуют такие факты «исключительной живучести» концепции ЭО, как [159]:
Формализация электроотрицательности на языке орбитальных свойств атомов и ионов делает её необходимым звеном полуэмпирического описания и моделирования химической связи.
ЭО позволяет наблюдать отчётливую периодичность и подчёркнутую контрастность свойств (электроположительные и электроотрицательные элементы, мягкие и жёсткие основания и кислоты), которая является основой природы химического взаимодействия.
В настоящее время существует десятки шкал ЭО, в основу расчётов которых положены различные свойства веществ и элементов, составляющих их. Значения ЭО разных шкал отличаются, но относительное расположение в ряду ЭО примерно одинаково. ЭО элемента зависит от многих факторов, в частности, от валентного состояния элемента, типа соединения, в которое он входит, но, тем не менее, это понятие необходимо для качественного объяснения свойств химической связи, химических соединений и материалов на их основе [40].
В работе [45] делается фундаментальный вывод о постоянстве ЭО в данном валентном состоянии «на начальный момент взаимодействия» и приводится следующая ее формулировка: «эта зависящая от природы и валентного состояния, присущая элементу способность притягивать электроны при образовании химической связи».
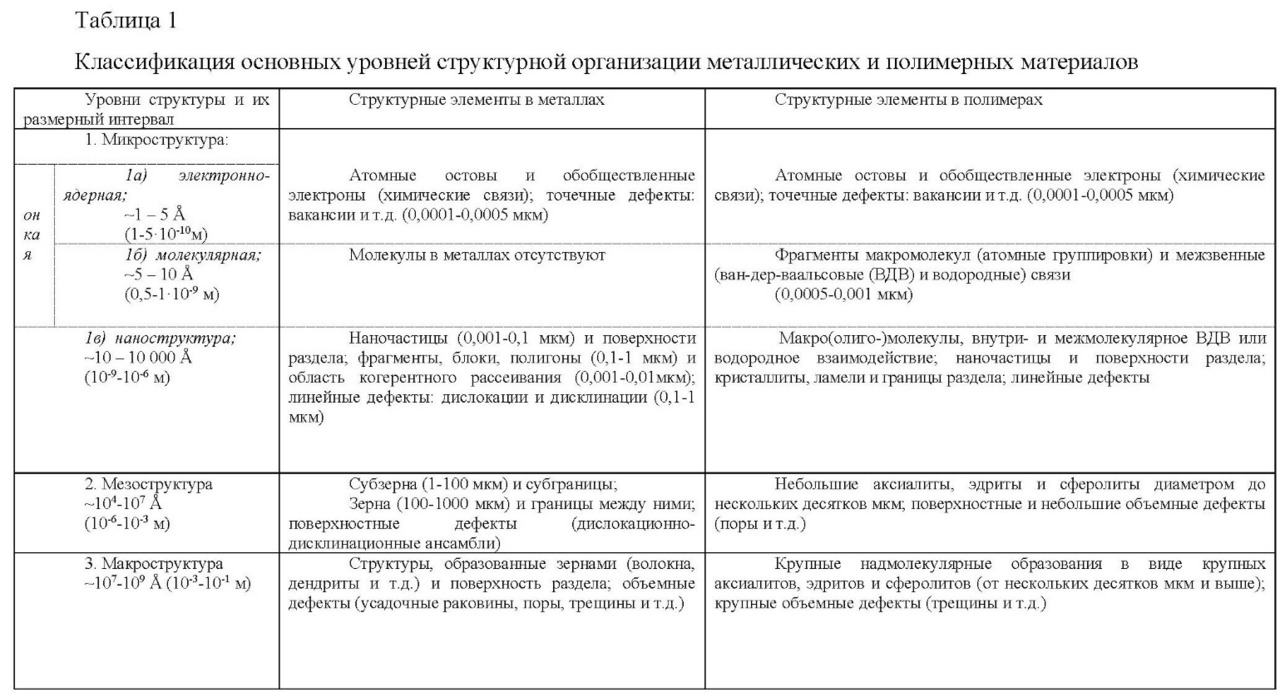
Полинг, предположив, что энергия связи равна теплоте, выделяемой при реакции, предложил использовать непосредственно тепловые эффекты Q, минуя данные об энергиях связи. Использование значений Q для расчётов основано на том, что большинство элементов в их стандартных состояниях содержат одинарные ковалентные связи.
Однако если же исходные элементы находятся в жидком или твёрдом состоянии, то кроме энергий ковалентных связей им присущ некоторый запас ВДВ энергии, которую шкала Полинга не учитывает. Полинг предложил в качестве первого приближения формулу вычисления ЭО элементов из тепловых эффектов [46]:
где nA и nB — числа атомов, образующих молекулу.
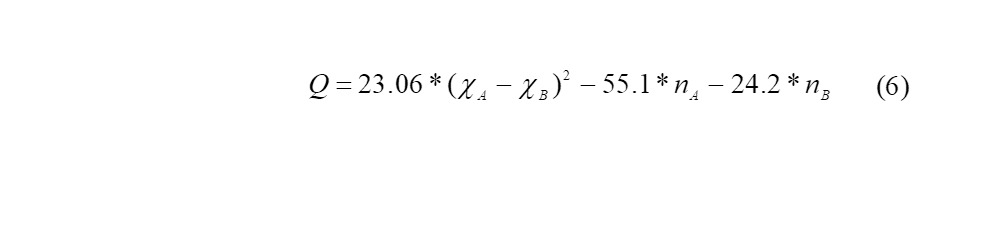
Таким образом, Полинг сформулировал зависимость полярности связи только от энергии химической связи, найденной из теплоты, выделяемой при реакции в виде термохимической концепции электроотрицательности атомов.
Вместе с тем, основная идея Полинга — зависимость энергии связи только от теплоты, выделяемой при реакции — требует корректировки. Очевидно, что энергия связи в немалой степени зависит и от её длины [46].
Вслед за Полингом термомеханические расчёты были произведены рядом авторов, из которых можно сделать вывод, что увеличение положительной валентности повышает ЭО атомов [47,48].
Другое направление расчёта ЭО — исходя из величин ковалентных радиусов (rК). Например, Оллред и Рохов [49,50] разработали альтернативный метод расчёта, исходя из эффективного заряда и rК атома:
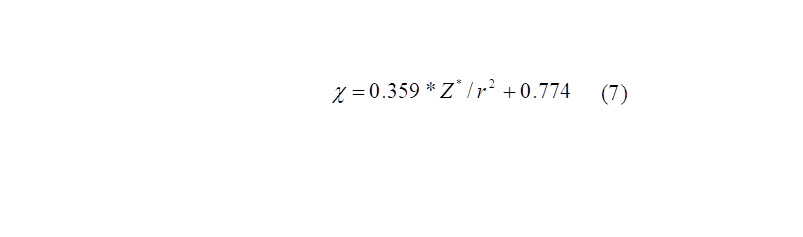
ЭО, как по Полингу, так и по Оллреду, как правило, безразмерные величины.
Пирсон [51,52] предложил шкалу абсолютной ЭО, которая определяется как среднее из первого потенциала ионизации и сродства к электрону для нейтрального атома. Обе последние величины были взяты Пирсоном в электрон-вольтах (эВ), следовательно, и значения абсолютной ЭО получились в электрон-вольтах, в то время как в других шкалах ЭО есть величины безразмерные [51,52].
Анализируя все известные на сегодня шкалы ЭО, можно заметить их недостатки. Оригинальная шкала Полинга ограничена валентными состояниями атомов с максимальной «нормальной» валентностью. Тем не менее, в пределах области своей применимости формальный подход Полинга является стройной логически замкнутой феноменологической теорией [45].
Сироткиным О. С. и д.р. [53] была разработана скорректированная шкала ЭО, лишенная недостатков шкал Полинга и Оллреда-Рохова, то есть было устранено присутствие элементов с одинаковыми значениями ЭО за счет использования не только ковалентных, но и металлических радиусов, а также других характеристик основных элементов ПС [53] (табл. 2).
1.5.2. Практическое использование электроотрицательностей

ЭО элементов традиционно используются для определения типа гетероядерной химической связи [54,55]. Их использование базируется на концепции поляризации химической связи, усиливающейся по мере увеличения разности ЭО элементов (ионов), образующих данное соединение. Эта концепция базируется на как будто весьма очевидном допущении — увеличение разности ЭО приводит к «перетягиванию» локализованного электронного облака связывающих электронов к более электроотрицательному элементу. Но этот подход не учитывает металлическую составляющую гетероядерной химической связи, которая в общем случае должна рассматриваться как ионно-ковалентно-металлическая. Необходимость учитывать «степень металличности» (СМ) в соединениях отметили Музер и Пирсон [56,57—59]. Однако в последующие годы на это обстоятельство обращали внимание лишь отдельные исследователи [51, 60, 61—64], тогда как подавляющее число авторов оставались на ортодоксальных поляризационных позициях рассмотрения ХСв, ограничивая ее вариации определением степени ионности (СИ) (степени ковалентности (СК)), считая ее прямо зависящей только от разности ЭО атомов (ионов), образующих то или иное определенное вещество.
Также от ЭО зависит такая фундаментальная характеристика ХСв, как ее СК, а, следовательно, прочность связи, тип структуры, особенности химического состава. ЭО в сочетании с другими факторами является решающей в формировании конкретных значений большинства физических и физико-химических свойств. СК возрастает при уменьшении разности ЭО соответствующих элементов [65].
С помощью ЭО вычисляются, например, такие характеристики, как температура Кюри [66]. Увеличение температуры Кюри по мере увеличения ЭО ионов объясняется усилением СК образуемой ими химической связи, а также их различий, связанных с числом валентных электронов с орбитальным квантовым числом l≥1 и интегралом перекрывания орбиталей этих электронов с валентными орбиталями кислорода [66]. Все это обеспечивает повышенную устойчивость таких связей к воздействию дестабилизирующих факторов, в частности, температуры. Установленные зависимости могут рассматриваться как основа для прогнозирования свойств и поиска новых высокотемпературных сегнетопьезокерамических материалов [67].
В 1991 году была предпринята попытка использовать концепцию ЭО Полинга применительно к анализу сверхпроводящих веществ [68].
Таким образом, из выше изложенного видно, что от ЭО зависит такая фундаментальная характеристика химической связи, как ее СК, а, следовательно, прочность связи, тип структуры, особенности химического состава. ЭО в сочетании с другими факторами является решающей в формировании конкретных значений большинства физических, физико-химических и физико-механических свойств. СК возрастает при уменьшении разности ЭО соответствующих элементов.
1.6. Расчет компонент химических связей
1.6.1. Гомоядерные (металло-ковалентные) химические связи
Законы движения микрочастиц в квантовой механике выражаются уравнением Шредингера. В квантово-механической теории одновременно развивались два разных метода приближённого решения уравнения Шредингера для случая молекул:
Метод валентных связей (ВС), который рассматривает волновую функцию, описывающую движение обобществлённой пары электронов с противоположно направленными спинами, образующих связь в молекуле.
Теория молекулярных орбиталей (МО), которая исходит из положения, что любая молекула характеризуется набором молекулярных орбиталей, охватывающих всю молекулу в целом и делокализованных между атомами. Обе теории в своих приближениях дают достаточно верные результаты. Каждая из этих теорий при описании имеет свои преимущества и недостатки [69]. Сначала квантово-механические расчеты производились с использованием метода МО, так как его расчётная схема удобна и универсальна. Затем стало укрепляться мнение, что локализованное описание более удобно с расчётной точки зрения. К тому же в ряде случаев открывались новые возможности для структурно-химического исследования отдельных взаимодействий, например, в изучении механизмов химических реакций, где локализованное описание облегчает идентификацию отдельных структурно выделенных стадий химического превращения [70]. В последнее время наблюдается сближение обоих методов.
Учёт резонансных структур вводит в метод ВС элементы делокализации; учёт конфигурационного взаимодействия в методе МО приводит к более локализованным структурам. Так в методе молекулярных орбиталей линейной комбинации атомных орбиталей (МО ЛКАО) упорядоченные по энергии МО образуют канонический номер орбиталей. Канонические МО можно преобразовывать в локализованные орбитали, применяя различные критерии локализации [71].
При квантово–механическом изучении структуры молекул широко применяют фрагментарный подход, когда молекула рассматривается как система связанных между собой фрагментов, в качестве которых выбираются такие химические частицы, которые сохраняют в основном свои свойства и геометрическую структуру в разных молекулах. Тогда образование химических связей между фрагментами описывается через взаимодействие фрагментальных орбиталей. Ещё одним методом получения информации о локализуемости электронов из электронной волновой функции, описывающих систему, является теория лоджий [71], согласно которой химическая связь представляет собой некоторую область общего объёма молекулы, окружающую два или более соседних атомных остова, в которых флуктуация числа электронов мала. В металлах кроме катионов, связанных «электронным газом», присутствуют электронейтральные атомы, которые могут объединяться частью «электронного облака» с определенной локализацией ЭП между ними и в результате непрерывного обмена валентных электронов, позволяющей образовывать в структуре металлических соединений связи между электронейтральными атомами. Это означает, что даже «чисто металлические связи» обладают некоторой СК [7]. Таким образом, можно сделать вывод, что большинство гомоядерных химической связи может одновременно характеризоваться определенной степенью локализации и делокализации, то есть не может рассматриваться как «чисто металлическая» или «чисто ковалентная». Можно вести речь лишь о соотношении локализации и делокализации, например, соотношении ковалентности и металличности [36,72], где для оценки СК или СМ соединения в качестве индивидуальной характеристики природы атома элемента возможно использования ЭО — Х или первого ПИ — I1 [36,72]. Отмечается, что увеличение суммарного атомного номера атомов соединения приводит к увеличению СМ связи [72]. При количественной характеристике типа химической связи в металлоковалентных гомосоединениях [36,72] предполагается, что СК пропорциональна ЭО (I1) атомов соединения. Отсюда следует, чтоэти формулы призваны учесть способность атомов отдавать свои электроны, то есть в них оценивается степень размытости электронного облака по оси перпендикулярной линии, соединяющей центры ядер химически связанных атомов.

В 1997 году эти формулы были уточнены Сироткиным О. С., Сироткиным Р. О., и Никифоровой Е. А. [73] путем введения в них коэффициента k, учитывающего природу элемента (s-, p-, d- и f-).
В дальнейшем было отмечено, что химический смысл металличности гомоядерной связи заключается в способности к смещению или делокализации обобществленных электронов в направлении перпендикулярном оси, соединяющие центры одинаковых ядер. Ее можно оценить [36,74,75] через степень отклонения ЭП обобществленных электронов по оси Х перпендикулярной линии, соединяющей центры ядер. Логично, что с увеличением металличности (в соответствии с уменьшением ЭО и I1 элемента) происходит увеличение этого отклонения или «степени размывания» обобществленной ЭП и ковалентной составляющей гомоядерной связи (то есть проявление тенденции к делокализации химической связи по оси Х, а, следовательно, и к росту её металличности).
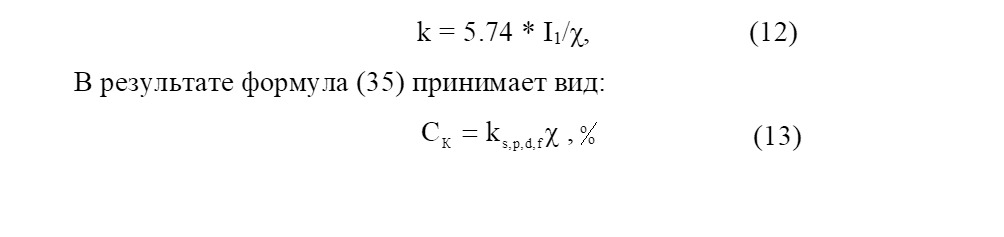
Авторами [36,74,75] были получены результаты по СМ (СК) различных гомосвязей в металлических, неметаллических низко и высокомолекулярных соединений на основе ЭО и первого ПИ — I1, и с помощью квантово-химических расчетов через момент первого порядка М1 (х).
На основе выражения для плотности электронного распределения вдоль оси X, вида молекулярных (Ψ) и атомных (φ) орбиталей, и расчета их составляющих авторами [74,75] была разработана программа на языке Basic и результатом расчетов с ее использованием явились графические зависимости P (x), которые не противоречат высказывавшемся ранее взглядам [73,76,77] на увеличение СМ гомосвязи при переходе внутри группы сверху вниз и общей взаимосвязи её величины с природой элементов образующих соответствующее химическое гомосоединение.
В качестве возможных конкретных количественных критериев металличности авторы предложили следующие два основных варианта.
Центр тяжести зарядовой плотности — момент первого порядка (M1 (x)):
При их сравнении выяснили, что «что M1 (x) наиболее адекватно описывает характер отклонения ЭП вдоль оси X при переходе от начала в конец» [74,75].
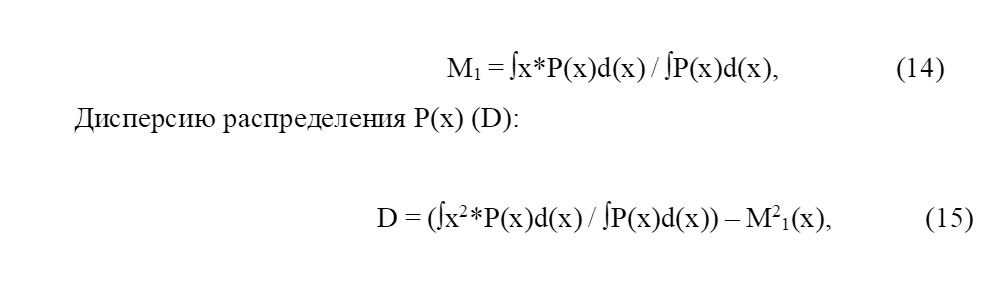
В работе [75] сделан вывод о том, что «традиционные химические подходы (основанные на использовании ЭО и первого ПИ) с учетом химической практики более адекватно описывают абсолютную величину и характер изменения СМ гомосвязей. Однако полученные в данной работе результаты позволили показать принципиальную возможность применения квантово-химических подходов к интерпретации ряда классических положений теории химической связи, в том числе, применительно к возможности прямой оценки металлической составляющей гомоядерного химического взаимодействия» [68].
Данные по СК связи гомополимеров позволили разобраться в природе гомосвязей на примере связи С-С [36].
На основе СМ и СК углерод-углеродных связей в различных по пространственной структуре карбоцепных полимерах проанализирована взаимосвязь этих характеристик с длиной и энергией соответствующих связей, а также со свойствами и мерностью макромолекулярных структур [78]. В зависимости от мерности полимеров различают одномерную линейную (карбин и карбен), двумерную слоистую (графит) и трехмерную (алмаз). В результате по этим значениям по формуле (35) можно оценить СК (СМ) для данных соединений. С увеличением мерности (структурной сложности) полимеров происходит увеличение величины ЭО атома углерода, СК связи С-С и МР между атомами углерода. Это ведет к снижению энергии связи. С увеличением СМ происходит укорачивание длины связи и соответствующие изменения физико-химических свойств полимеров. Также с увеличением мерности полимера возрастает его плотность [78].
Бацанов С.С предложил «метод оценки поляризующего действия атомов и количественную оценку металличности связи в кристаллических соединениях» [79] и на их основе рассчитал «молекулярные рефракции… галогенидов, оксидов и халькогенидов с точностью в несколько раз лучшей, чем при ионном подходе» [79]. Мольная рефракция учитывает действие поляризуемости атомов, взаимодействие связей и приводящее к увеличению электронной поляризуемости; а также поправку из-за наличия в структуре металлических связей (ΔRm). Металличность связей (m) автор определяет как «долю ковалентных электронов, образующих связи А — А в соединении АВ» и рассчитывает по формуле
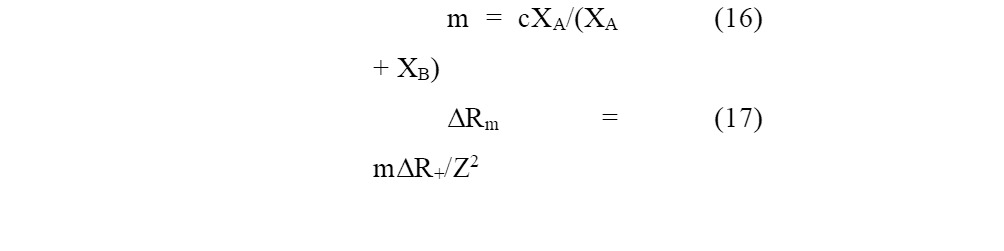
где с — ковалентность связи; Х — кристаллические ЭО атомов А и В с эффективными зарядами ±i; ΔR+ — рефракция катиона; Z — валентность атома.
Все это дает основание использовать рефрактометрию для изучения структуры соединений [79].
1.6.2. Гетероядерные (металло-ионно-ковалентные) химические связи
Природу ионной связи чаще объясняют с позиций электростатического взаимодействия ионов [12]. Переход электронов между атомами А и В, резко отличающихся по ЭО, превращает атомы в противоположно заряженные ионы. Между ионами А и В возникает электростатическое взаимодействие, которое приводит к образованию молекулы. Установлено, что ионная связь в чистом виде не встречается. Даже при взаимодействии щелочных металлов с галогенами модно вести речь лишь о преимущественном проявлении ионной связи химических соединений.
Молекулы, состоящие из неодинаковых атомов, характеризуются смещением электронных пар к более электроотрицательному атому. С точки зрения молекулярных орбиталей [40], в гетероядерных двухатомных молекулах энергия атомных орбиталей и их относительный вклад в молекулярные обитали различен. Связывающие орбитали по энергии ближе к орбиталям более электроотрицательного атома, а разрыхляющие — к орбиталям менее электроотрицательного атома. Различие в энергии исходных АО определяет полярность (ионность) связи. Ионность связи для молекулы рассчитывается [89] из экспериментальных значений дипольных моментов:
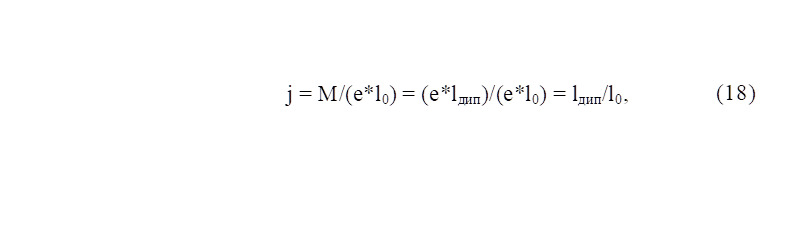
где М — дипольный момент молекулы; е — заряд электрона; lдип — расстояние между ЦТ положительных и отрицательных зарядов в молекуле; l0 — межъядерное расстояние в молекуле (длина связи).
Эта формула имеет свои недостатки:
Ионность с изменением МР изменяется не линейно. Чем больше является ионной связь, тем труднее происходит сжатие оболочек.
Исходя из этой формулы, можно было бы предположить, что молекула, в которой положительный и отрицательный заряды раздвинуты на расстояние, будет чисто ионной. Но в чисто ионной молекуле силы притяжения значительно больше, чем в полярной молекуле, то есть длина связи уменьшается по сравнению с [80]. Тем не менее, эта формула стала основной для вычисления Полингом ионности связи.
Ионность связи, выраженная в процентах, называется степенью ионности i. Также i связи рассчитывается по формуле
где q — эффективный заряд ядра; e — заряд электрона; μэксп — экспериментальный дипольный момент молекулы; μион — гипотетический дипольный момент молекулы СИ связи, как мера её полярности, зависит от различия в электроотрицательности атомов, участвующих в образовании связи [81]. Опираясь на исходные значения галогенидов, рассчитанных по значениям дипольных моментов, Полинг построил эту эмпирическую зависимость в следующем виде:
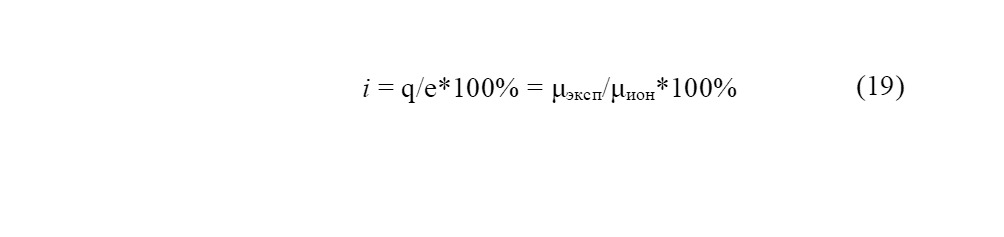
где EA, B — сродство к электрону атомов А и В

В работах Сандерсена [44] были описаны не только методы расчётов отношения стабильности (ОС) атомов и молекул, но и способы вычисления ионного характера связей. Согласно Сандерсену, ОС или ЭО любого атома в молекуле будет равняться ОС остальных атомов, а значит и всей молекулы в целом. Однако, до сих пор, уравнение, предложенное Полингом для расчёта ионности связи, используется наиболее широко.
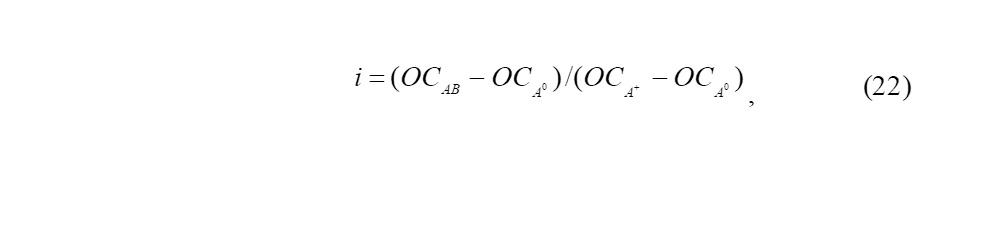

Сладков И. Б. [83] в 1987 году предложил простой способ оценки полярности и ионности жидкостей с помощью параметра, вычисленного из их термодинамических характеристик в точке кипения. Выводы о степени полярности вещества могут быть сделаны из численных характеристик его дипольного момента, потенциала ионизации и сродства к электрону. Также они зависят от размеров и конфигурации молекулы. Автор для характеристики степени полярности предложил использовать «параметр, характеризующий отклонение межмолекулярного взаимодействия в конкретном веществе от наиболее простого, реализуемого в неполярных сферически-симметричных молекулах» (в дальнейшем ψ-фактор) и рассчитал его через Ткип и Vкип (мольный объем в точке кипения). Он обнаружил корреляцию между ψ-фактором и дипольным моментом в галогеноводородах [83]. Также наблюдается уменьшение ψ-фактора при переходе от более ионного соединения к менее ионному (неполярному) в ряду «галогенидов элементов, расположенных в одном ряду периодической системы… с уменьшением атомной массы элемента» [83]. С уменьшением полярности в ряду галогенидов от фторида к иодиду ψ-фактор уменьшается. На основе этих данных можно выявить 3 группы веществ с различными значениями ψ-фактора: Для неполярных и слабо полярных жидкостей он меньше 0.05; для умеренно полярных его значение составляет 0.05 — 0.1 и для сильно полярных ψ больше 0.1. Информация о СИ также можно оценить исходя из значений ψ-фактора: для жидкостей ионного характера нижняя граница ψ-фактора составляет 0.3, а для молекулярных жидкостей это значение не должно превышать 0.25 [83].
По мнению О. С. Сироткина химическую связь следует рассматривать как наложение ковалентного, ионного и металлического состояний [14]. В итоге химическая связь, определяемая характером распределения обобществленных электронов в межъядерном пространстве, должна рассматриваться на качественном уровне, как результат наложения всех трех компонент химической связи друг на друга [14], а характер их локализации-делокализации можно описать в общем виде уравнением суммарной волновой функции.
где С1, С2 и С3 — коэффициенты, определяющие долю ковалентной, металлической и ионной составляющих связи, которые в сумме равны единице или 100%. По сути уравнение (23) объединяет классические и квантово-механические подходы для описания реальных химических связей в рамках единой универсальной модели [14]. В случае гомоядерных связей С3 = 0 и, следовательно, уравнение (23) упрощается за счет ликвидации последнего его члена [14].
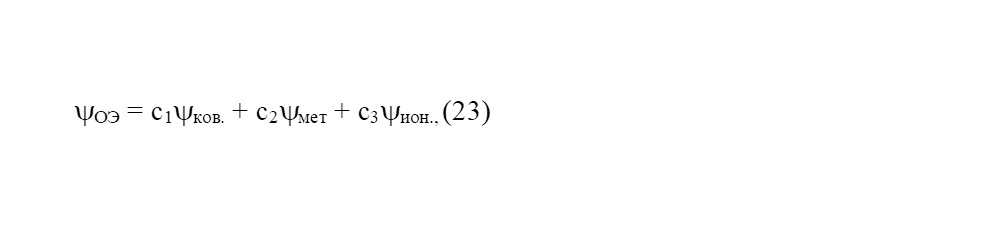
В результате вышесказанного возникает необходимость учета вклада металлической компоненты в гетероядерное взаимодействие и оценки степени металличности в нем. В качестве варианта решения задачи количественной оценки CМ гетероядерного взаимодействия k заменяется на kcp, a χ на χср. Определение значения ЭО через точку, местоположение которой соответствует центру тяжести электронной плотности (χср) на оси X, позволяет в дальнейшем использовать его для определения CК. В результате формула (23) приобретает вид:
После вычисления всех трех компонент химической связи по формулам (24) — (27) определяем сумму составляющих гетероядерной связи, принимаем ее за 100%, и рассчитываем приведенные степени ионности, ковалентности и металличности гетероядерной химической биядерной связи по формулам [53]:
Бесплатный фрагмент закончился.
Купите книгу, чтобы продолжить чтение.